
BS-Seeker2
A versatile aligning pipeline for bisulfite sequencing data
Documents | Release Log | Q&A | Source Code | Tutorial ㊥ | Jan. 3rd, 2018 Updated
DOWNLOAD newest version !
Archived versions:
NOTICE The newest version (V2.1.0) has been tested on python v2.7 and pysam v0.9+/v0.8; and python v2.7 and pysam v0.75 For BS-Seeker2 older versions, if you notice pysam related problem, please try a older pysam version, such as pysam v0.6.
News A new software package CGmapTools is now recommended, for the seamless downstream analysis following BS-Seeker2.
Features
- WGBS / RRBS
- single-end / paired-end
- Local / gapped alignment
- Galaxy
- Input formats
- Fasta
- Fastq
- qseq
- pure sequence
- Output formats
- BAM
- SAM
- BS_Seeker
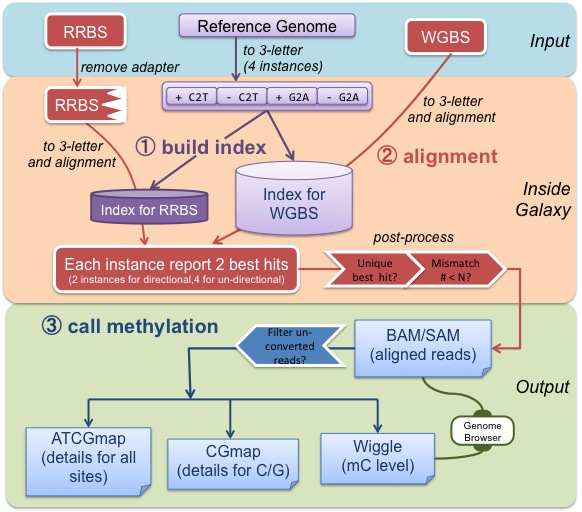
"How can I use BS-Seeker2?"
- Command line
- Galaxy
- UCLA : Hoffman2 or MCDB using your own account.
- Apply MCDB galaxy account by emailing Weihong Yan and tell that you love "BS-Seeker2"!
- Galaxy Toolshed
Read requirements . Download newest version . Run BS-Seeker2 following the documents.
"Can you give me some examples to start with BS-Seeker2?"
Sure! But you should know that our BS-Seeker2 is very versatile, and you can make a better use of it by reading more about the help information.
1. Build the index
# WGBS, using bowtie2 (path of bowtie should be included in $PATH)
python bs_seeker2-build.py -f genome.fa
# RRBS, using bowtie
python bs_seeker2-build.py -f genome.fa --aligner=bowtie -p ~/install/bowtie/ -r
# RRBS, with fragment lengths ranging [40bp, 400bp]
python bs_seeker2-build.py -f genome.fa -r -l 40 -u 400
# RRBS for double-enzyme : MspI (C'CGG) and ApeKI (G'CWGC)
python bs_seeker2-build.py -f genome.fa -r -c C-CGG,G-CWGC
2. Map the reads
# WGBS, FASTA, bowtie2 (local alignment), allowing 3 mismatches
python bs_seeker2-align.py -i WGBS.fa -m 3 --aligner=bowtie2 -o WGBS.bam -f bam -g genome.fa
# RRBS, FASTQ, bowtie, output as SAM
python bs_seeker2-align.py -i RRBS.fq --aligner=bowtie -o RRBS.sam -f sam -g genome.fa -r -a adapter.txt
# RRBS, QSEQ, bowtie2 (end-to-end alignment), fragment lengths ranging [40bp, 400bp]
python bs_seeker2-align.py -i RRBS.qseq --aligner=bowtie2 --bt2--end-to-end -o RRBS.bam -f bam -g genome.fa -r --low=40 --up=400 -a adapter.txt
# Paired-end, QSEQ, bowtie, report concordant reads, and remap the unmapped reads in single-end mode
# Output the unmapped reads in PE mode
bs_seeker2-align.py -1 FN1 -2 FN2 -g genome.fa -o PE.bam -u unmapped
# Map the unmapped reads in mate 1
bs_seeker2-align.py -i unmapped_1.fa -g genome.fa -o unmapped_1.bam
# Convert the unmapped reads in mate 2 to their reverse complementaries
Antisense.py -i unmapped_2.fa -o unmapped_2_antisense.fa
# Map the unmapped reads in mate 2
bs_seeker2-align.py -i unmapped_2_antisense.fa -g mm9_phage.fa -o unmapped_2.bam
# Merge all the mapped results
samtools merge merge.bam PE.bam unmapped_[12].bam
3. Call the methylation levels
# WGBS, bowtie
python bs_seeker2-call_methylation.py -i WGBS.bam -o output --db /path/to/BSseeker2/bs_utils/reference_genomes/genome.fa_bowtie/
# RRBS, bowtie2, fragment lengths ranging [40bp, 400bp]
python bs_seeker2-call_methylation.py -i RRBS.bam -o output --db /path/to/BSseeker2/bs_utils/reference_genomes/genome.fa_rrbs_40_400_bowtie2/
# RRBS, bowtie2, removing the incomplete bisulfite converted reads
python bs_seeker2-call_methylation.py -x -i RRBS.bam -o output --db /path/to/BSseeker2/bs_utils/reference_genomes/genome.fa_rrbs_40_500_bowtie2/
# RRBS, only show sites covered by at least 10 reads in WIG file
python bs_seeker2-call_methylation.py -r 10 -i RRBS.bam -o output --db /path/to/BSseeker2/bs_utils/reference_genomes/genome.fa_rrbs_40_500_bowtie2/
Need more information?
- Document of BS-Seeker2 .
- Question and Answer (Q&A).
- Output formats o of BS-Seeker2.
- The LOGS for releases
Do a fast test using BS-Seeker2?
We provide a small Data set for fast testing purpose, which is in the "test_data" folder. Or you can download them from here .
Contact:
Please contact Weilong Guo if you have any question.
Email : guoweilong[aite]126.com ( Please replace "[aite]" by @ )
Citation:
Guo W, Fiziev P, Yan W, Cokus S, Sun X, Zhang MQ, Chen P, Pellegrini M. (2013). BS-Seeker2: a versatile aligning pipeline for bisulfite sequencing data. BMC Genomics, 14(1), 774. doi:10.1186/1471-2164-14-774
Datasets for paper:
- DS1_WGBS_simu_perfect.fa
- DS2_PE_simu_perfect_end1.fa and DS2_PE_simu_perfect_end2.fa
- DS3_RRBS_simu_perfect.fa
- DS5_PE_simu_error_end1.fa and DS5_PE_simu_error_end2.fa
- DS4_WGBS_simu_error.fa
- DS6_RRBS_simu_error.fa
- DS7_WGBS_real.fa
- DS8_PE_real_end1.fa and DS8_PE_real_end2.fa
- DS9_RRBS_real.fa
- DS10_Phage_spikein.qseq
- DS11_Sample_A.qseq
- DS12_Sample_B.qseq
Other link :
Who are using BS-Seeker2?
